The Beran Group
Department of Chemistry, University of California at Riverside
Accurate quantum chemistry in the condensed phase.
Developing practical new theoretical models for describing
non-covalent interactions and predicting the structures and properties
of molecular crystals.
List of Publications
Last updated December 2025. Google Scholar. Review Articles, Editorials and Press.2025
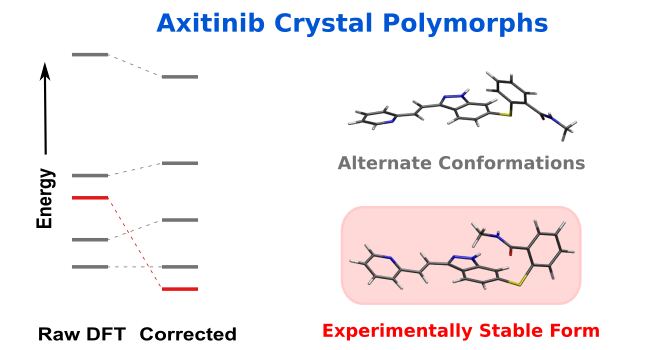
[121] "From polymorphs to cocrystals and salts: successfully predicting axitinib's challenging crystal forms." G. Beran. Chem. Sci. 16, 23026-23037 (2025). DOI: 10.1039/D5SC06271C
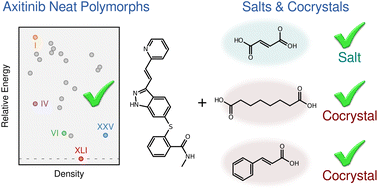
Featured in: Chemistry World
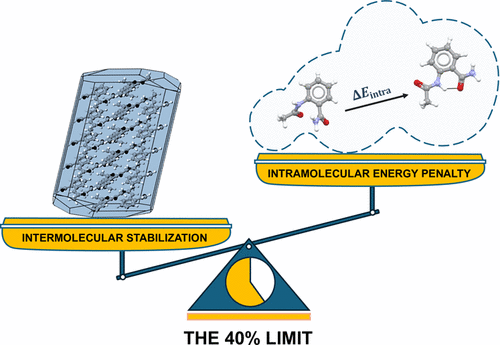
[120] "Lattice Energy Partitions in Crystals of Flexible Molecules and the 40% Limit." A. Chattopadhyay, A. Hill, S. Wright, G. Beran, and A. Cruz-Cabeza. J. Am. Chem. Soc. 147, 39192-39203 (2025). DOI: 10.1021/jacs.5c09997
[119] "Photomechanical Anthracenophane Crystals: Theory, Experiment, and Composite Actuator Performance." K. Lam, P. Molina-Portillo, V. Carta, T. Nishiuchi, M. Ticknor, R. Hayward, R. Al-Kaysi, T. Kubo, G. Beran, and C. Bardeen. Cryst. Growth. Des. 25, 8089-8099 (2025). DOI: 10.1021/acs.cgd.5c00851
[118] "Radical-Initiated Photodissociation at Tryptophan Residues within Intact Proteins in the Gas Phase." L. He, A. Purcell, G. Beran, Y. Zhu, K. Coronado, S. Mann, W. Harman, and R. Julian. J. Am. Chem. Soc. 147, 28583-28588 (2025). DOI: 10.1021/jacs.5c07784
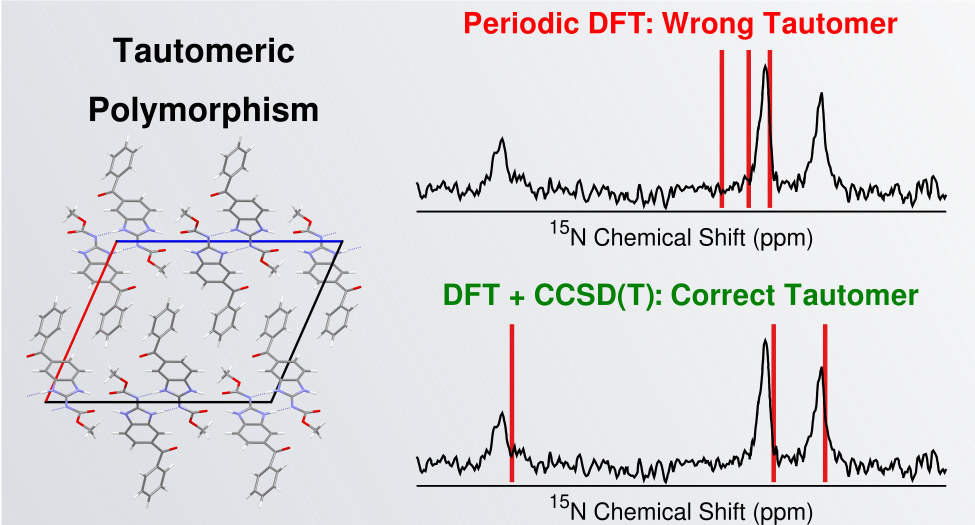
[117] "Taming tautomerism in organic crystal structure prediction." C. Perry, S. Ramos, M. Phelps, L. Mueller, and G. Beran. J. Am. Chem. Soc. 147, 26865-26876 (2025). DOI: 10.1021/jacs.5c08442
 [116] "Predicting solid-state NMR observables via machine learning."
P. Unzueta and G. Beran.
Modern NMR Crystallography: Concepts and Application, 224-255 (2025).
DOI: 10.1039/9781837673179-00224
[116] "Predicting solid-state NMR observables via machine learning."
P. Unzueta and G. Beran.
Modern NMR Crystallography: Concepts and Application, 224-255 (2025).
DOI: 10.1039/9781837673179-00224
[115] "Investigating cooperative reactivity in photomechanical crystals using first-principles density functional theory." C. Perry and G. Beran. Cryst. Growth Des. 25, 2561-2571 (2025). DOI: 10.1021/acs.cgd.5c00071
[114] "Distinctive Photomechanical Shape Change of p-Phenylenediacrylic Acid Dimethyl Ester Single Crystals Induced By Spatially Heterogeneous Photoreaction." D. Kitagawa, R Tomoda, S. Ramos, G. Beran, C. Bardeen, and S. Kobatake Angew. Chem. Int. Ed. 64, e202420243 (2025). DOI: 10.1002/anie.202420243 Selected as a "Hot Paper" by the journal editors.

2024
[113] "Using Chemical Substitution to Engineer Photomechanical Cinnamalmalononitrile Crystals." T. Gately, C. Perry, S. Weiss, K. Lam, I. Islam, M. Almitiri, V. Carta, G. Beran, and C. Bardeen. Cryst. Growth Des. 24, 9544-9555 (2024). DOI: 10.1021/acs.cgd.4c01033
[112] "The Seventh Blind Test of Crystal Structure Prediction:
Structure Ranking Methods." L. Hunnisett et al.
Acta
Cryst. B 80, 548-574 (2024). DOI:
10.1107/S2052520624008679
Official report on Structure Ranking from the 7th Blind Test of Crystal Structure Prediction.
[111] "Contrasting conformational behaviors of molecules XXXI
and XXXII in the seventh blind test of crystal structure
prediction." G. Beran, C. Cook, and P. Unzueta.
Acta
Cryst. B 80, 606-619 (2024). DOI: 10.1107/S2052520624005043
Report on our group's work in the 7th Blind Test of Crystal Structure Prediction.
[110] "Light-Triggered Rolling and Unrolling of Molecular Crystal Microsheets." P. Ghate, C. Perry, V. Carta, I. Bushnak, Y. Almuallem, G. Beran, C. Bardeen, and R. Al-Kaysi. Cryst. Growth Des. 24, 7695-7703 (2024). DOI: 10.1021/acs.cgd.4c00996
[109] "The interplay of density functional selection and
crystal structure for accurate NMR chemical shift predictions."
S. Ramos, L. Mueller, and G. Beran.
Faraday
Disc. 255, 119-142 (2025). DOI: 10.1039/D4FD00072B
See also the associated discussions:
- "Big data and simulations in NMR crystallography: general discussion." S. Ashbrook et al, Faraday Disc. 255, 159-191 (2025). DOI: 10.1039/d4fd90054e
- "Challenges and opportunities for NMR calculations: general discussion." S. Ashbrook et al, Faraday Disc. 255, 288-310 (2025). DOI: 10.1039/d4fd90055c
2023
[108] "Improved Description of Intra- and Intermolecular Interactions Through Dispersion-Corrected Second-Order Møller-Plesset Perturbation Theory." G. Beran, C. Greenwell, C. Cook, and J. Rezac. Acc. Chem. Res. 56, 3525-3534 (2023). DOI: 10.1021/acs.accounts.3c00578

[107] Perspective: "Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials." G. Beran. Chem. Sci. 14, 13290-13312 (2023). DOI: 10.1039/D3SC03903J Open Access.
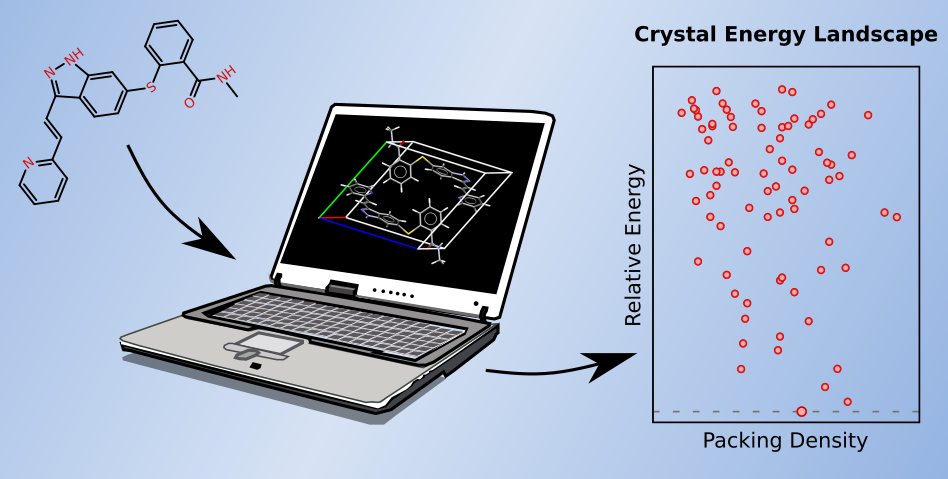
[106] "Understanding the impact of halogenation on the crystalline photomechanical response properties of 9-anthracene carboxylic acid from first-principles" C. Perry and G. Beran. Cryst. Growth Des. 23, 8352-8360 (2023). DOI: 10.1021/acs.cgd.3c00989
[105] "Organic Crystal Packing Is Key to Determining the Photomechanical Response." C. Cook, C. Perry, and G. Beran. J. Phys. Chem. Lett. 14, 6823-6831 (2023). DOI: 10.1021/acs.jpclett.3c01676
[104] "Do models beyond hybrid density functionals increase the agreement with experiment for predicted NMR chemical shifts or electric field gradient tensors in organic solids?" R. Iuliucci, J. Hartman, and G. Beran. J. Phys. Chem. A 127, 2846-2858 (2023). DOI: 10.1021/acs.jpca.2c07657 Invited contribution to the special issue for MQM 2022.
[103] "Polymorphs, Solvatomorphs, Hydrate and Perhydrate of Dabrafenib." S. Rai, A. Gunnam, G. Beran, J. Kaduk, and A. Nangia. Cryst. Growth Des. 23, 1179-1188 (2023). DOI: 10.1021/acs.cgd.2c01289
[102] "A theoretical framework for the design of molecular crystal engines." C. Cook, W. Li, B. Lui, T. Gately, R. Al-Kaysi, L. Mueller, C. Bardeen, and G. Beran. Chem. Sci. 14, 937-949 (2023). DOI: 10.1039/D2SC05549J Open Access.

2022
[101] "Effect of fluorination on polymorphism and photomechanical properties of cinnamalmalononitrile crystals." T. Gately, C. Cook, R. Almuzarie, I. Islam, Z. Gardner, R. Iuliucci, R. Al-Kaysi, G. Beran, and C. Bardeen. Cryst. Growth Des. 22, 7298-7307 (2022). DOI: 10.1021/acs.cgd.2c00930
[100] "Correcting π-delocalization errors in conformational energies using density-corrected DFT, with application to crystal polymorphs." B. Rana, G. Beran, and J. Herbert. Mol. Phys. 121, e2138789 (2023). DOI: 10.1080/00268976.2022.2138789. Nick Besley Memorial Issue. Open Access.
[99] "The interplay of intra- and intermolecular errors in modeling conformational polymorphs." G. Beran, S. Wright, C. Greenwell, and A. Cruz-Cabeza. J. Chem. Phys. 156, 104112 (2022). DOI: 10.1063/5.0088027
[98] "Spin-component-scaled and dispersion-corrected second-order Møller-Plesset perturbation theory: A path toward chemical accuracy." C. Greenwell, J. Rezac, and G. Beran. Phys. Chem. Chem. Phys. 24, 3695-3712 (2022). DOI: 10.1039/D1CP04922D
[97] "How many more polymorphs of ROY remain undiscovered?" G. Beran, I. Sugden, C. Greenwell, D. Bowskill, C. Pantelides, and C. Adjiman. Chem. Sci. 13, 1288-1297 (2022). DOI: 10.1039/D1SC06074K Open Access.

Featured in: ChemSci Pick of the Week 1/19/22
2021
[96] "Effect of halogen substitution on energies and dynamics of reversible photomechanical crystals based on 9-anthracenecarboxylic acid." T. Gately, W. Sontising, C. Easley, I. Islam, R. Al-Kaysi, G. Beran, and C. Bardeen. CrystEngComm 23, 5931-5943 (2021). DOI: 10.1039/D1CE00846C
[95] "Modeling the α- and β-resorcinol phase boundary via combination of density functional theory and density functional tight-binding." C. Cook, J. McKinley, and G. Beran. J. Chem. Phys 154, 134109 (2021). DOI: 10.1063/5.0044385
[94] "Modeling small structural and environmental differences in solids with 15-N NMR chemical shift tensors." J. Harper, L. Wang, A. Elliott, S. Moore, G. Beran, and J. Hartman. ChemPhysChem 22, 1008-1017 (2021). DOI: 10.1002/cphc.202000985
[93] "Rubrene untwisted: common density functional theory calculations overestimate its deviant tendencies." C. Greenwell and G. Beran. J. Mater. Chem. C 8, 2848-2857 (2021). DOI: 10.1039/D0TC05463A
[92] "Predicting Density Functional Theory-Quality Nuclear Magnetic Resonance Chemical Shifts via Δ-Machine Learning." P. Unzueta, C. Greenwell, and G. Beran. J. Chem. Theory Comput. 17, 826-840 (2021). DOI: 10.1021/acs.jctc.0c00979
[91] "Bridging Photochemistry and Photomechanics with NMR Crystallography: the Molecular Basis for the Macroscopic Expansion of an Anthracene Ester Nanorod." K. Chalek, X. Dong, F. Tong, R. Kudla, L. Zhu, A. Gill, W. Xu, C. Yan, J. Hartman, A. Magalhaes, R. Al-Kaysi, R. Hayward. R. Hooley, G. Beran, C. Bardeen, and L. Mueller. Chem. Sci 12, 453-463 (2021). DOI: 10.1039/D0SC05118G Open Access.
2020
[90] "Reduced-cost supercell approach for computing accurate phonon density of states in organic crystals." C. Cook and G. Beran. J. Chem. Phys. 153, 224105 (2020). DOI: 10.1063/5.0032649
[89] "Polarizable continuum models provide an effective electrostatic embedding model for fragment-based chemical shift prediction in challenging systems." P. Unzueta and G. Beran. J. Comp. Chem. 41, 2251-2265 (2020). DOI: 10.1002/jcc.26388
[88] Communication: "Inaccurate conformational energies still hinder crystal structure prediction in flexible organic molecules." C. Greenwell and G. Beran. Cryst. Growth Des. 20, 4875-4881 (2020). DOI: 10.1021/acs.cgd.0c00676 Selected for ACS Editors' Choice.
[87] "Combining crystal structure prediction and simulated spectroscopy in pursuit of the unknown nitrogen phase ζ crystal structure." W. Sontising and G. Beran. Phys. Rev. Mater. 4, 063601 (2020). DOI: 10.1103/PhysRevMaterials.4.063601
[86] "Overcoming the difficulties of predicting conformational polymorph energetics in molecular crystals via correlated wavefunction methods." C. Greenwell, J. McKinley, P. Zhang, Q. Zeng, G. Sun, B. Li, S. Wen, and G. Beran. Chem. Sci. 11, 2200-2214 (2020). DOI: 10.1039/C9SC05689K Open Access.

2019
[85] "Calculating Nuclear Magnetic Resonance Chemical Shifts from Density Functional Theory: A Primer." G. Beran. eMagRes. 8, 215-226 (2019).
[84] "Theoretical assessment of the structure and stability of the λ phase of nitrogen." W. Sontising and G. Beran. Phys. Rev. Mater. 3, 095002 (2019). DOI: 10.1103/PhysRevMaterials.3.095002
[83] "Improving predicted nuclear magnetic resonance chemical shifts using the quasi-harmonic approximation." J. McKinley and G. Beran. J. Chem. Theory Comput. 15, 5259-5274 (2019). DOI: 10.1021/acs.jctc.9b00481
[82] "Improving the accuracy of solid-state nuclear magnetic resonance chemical shift prediction with a simple molecular correction." M. Dracinsky, P. Unzueta, and G. Beran. Phys. Chem. Chem. Phys. 21, 14992-15000 (2019). DOI: 10.1039/C9CP01666J
[81] "Towards reliable ab initio sublimation pressures for organic molecular crystals – Are we there yet?" C. Cervinka and G. Beran. Phys. Chem. Chem. Phys. 21, 14799-14810 (2019). DOI: 10.1039/C9CP01572H
[80] "Reduced computational cost of polarizable force fields by a modification of the always stable predictor-corrector." D. Nocito and G. Beran. J. Chem. Phys. 150, 151103 (2019). DOI: 10.1063/1.5092133
[79] "Solid state photodimerization of 9-tert-butyl anthracene ester produces an exceptionally metastable polymorph according to first-principles calculations." G. Beran. CrystEngComm 21, 758-764 (2019). DOI: 10.1039/C8CE01985A. Selected for the Editor's collection on Polymorphism in Molecular Crystals.
2018
[78] "Accurate 13-C and 15-N molecular crystal chemical shielding tensors from fragment-based electronic structure theory." J. Hartman and G. Beran. Solid State Nucl. Magn. Res. 96, 10-18 (2018). DOI: 10.1016/j.ssnmr.2018.09.003
[77] "Small Structural Variations Have Large Effects on the Assembly Properties and Spin State of Room Temperature High Spin Fe(II) Iminopyridine Cages." T. Miller, L. Holloway, P. Nye, Y. Lyon, G. Beran, W. Harman, R. Julian, and R. Hooley. Inorg. Chem. 57, 13386-13396 (2018). DOI: 10.1021/acs.inorgchem.8b01973
[76] "Accurate non-covalent interactions via dispersion-corrected second-order Møller-Plesset perturbation theory." J. Rezac, C. Greenwell, and G. Beran. J. Chem. Theory Comput. 14, 4711-4721 (2018). DOI: 10.1021/acs.jctc.8b00548
[75] "Massively Parallel Implementation of Divide-and-Conquer Jacobi Iterations Using Particle-Mesh Ewald for Force Field Polarization." D. Nocito and G. Beran. J. Chem. Theory Comput. 14, 3633-3642 (2018). DOI: 10.1021/acs.jctc.8b00328
[74] "Dipole Effects on Electron Transfer are Enormous." M. Krzeszewski, E. Espinoza, C. Cervinka, J. Derr, J. Clark, D. Borchardt, G. Beran, D. Gryko, and V. Vullev. Angew. Chem. Int. Ed., 57, 12365-12369 (2018). DOI: 10.1002/anie.201802637. Selected as a "Hot Paper" by the journal editors.
[73] "Ab initio prediction of the polymorph phase diagram for crystalline methanol." C. Cervinka and G. Beran. Chem. Sci. 9, 4622-4629 (2018). DOI: 10.1039/C8SC01237G Open Access.
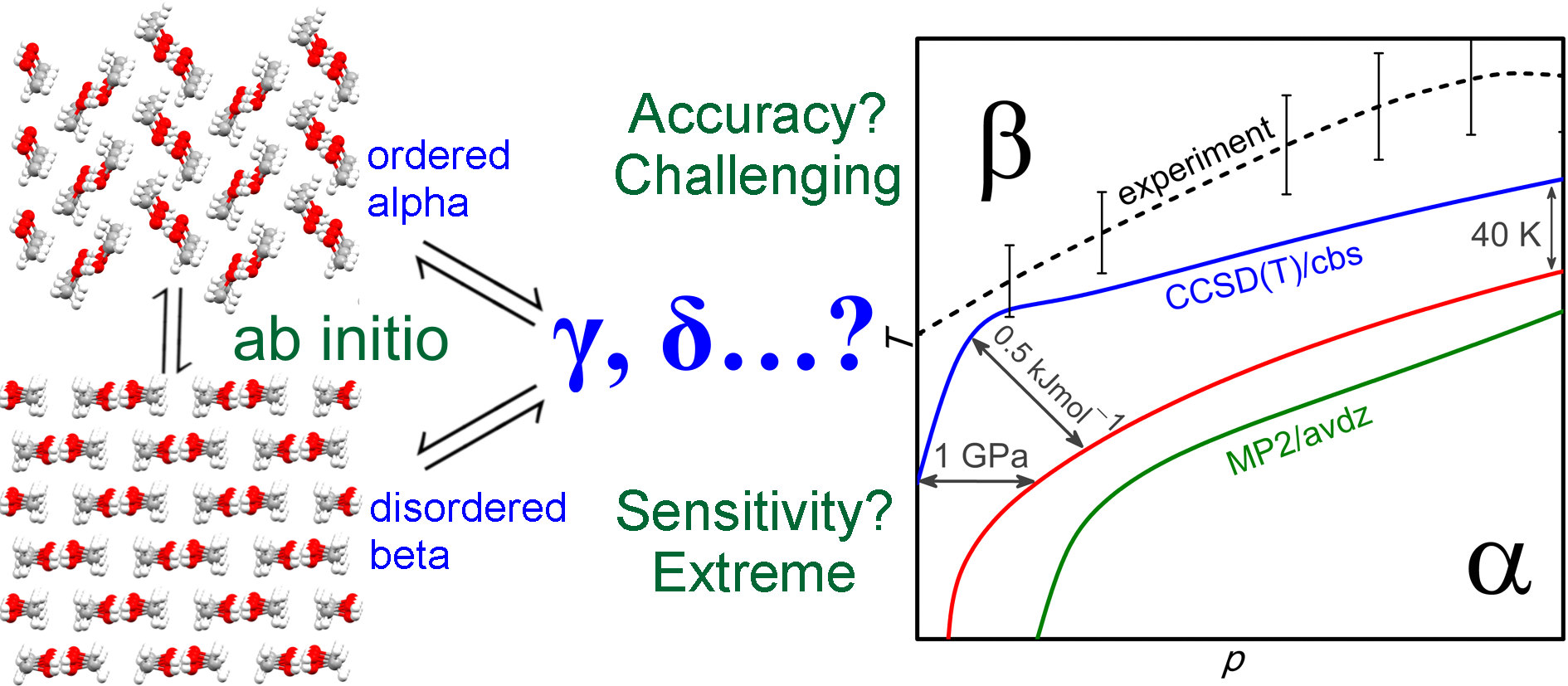
Featured in: Chemistry World,
ChemSci Pick of the Week 5/2/18, and
Inside UCR.
[72] "A Springloaded Metal-Ligand Mesocate Allows Access to Trapped Intermediates of Self-Assembly." P. Bogie, L. Holloway, Y. Lyon, N. Onishi, G. Beran, R. Julian, and R. Hooley. Inorg. Chem. 57, 4155-4163 (2018). DOI: 10.1021/acs.inorgchem.8b00370
[71] "Identifying pragmatic quasi-harmonic electronic structure
approaches for modeling molecular crystal thermal expansion."
J. McKinley and G. Beran.
Faraday
Disc. 211, 181-207 (2018). DOI: 10.1039/C8FD00048D
See also the associated discussions:
- "Structure searching methods: General discussion." M. Addicoat et al, Faraday Disc. 211, 133-180 (2018). DOI: 10.1039/C8FD90030B
- "Crystal structure evaluation: calculating relative stabilities and other criteria: general discussion." M. Addicoat et al, Faraday Disc. 211, 325-381 (2018). DOI: 10.1039/C8FD90031K
2017
[70] "Improved Electrostatic Embedding for Fragment-Based Chemical Shift Calculations in Molecular Crystals." J. Hartman, A. Balaji, and G. Beran. J. Chem. Theory Comput. 13, 6043-6051 (2017). DOI: 10.1021/acs.jctc.7b00677
[69] "Ab Initio Thermodynamic Properties and Their Uncertainties for Crystalline α-Methanol." C. Cervinka and G. Beran. Phys. Chem. Chem. Phys. 19, 29940-29953 (2017). DOI: 10.1039/C7CP06605H
[68] "Theoretical Predictions Suggest Carbon Dioxide Phases III and VII are Identical." W. Sontising, Y. Heit, J. McKinley, and G. Beran. Chem. Sci. 8, 7374-7382 (2017). DOI: 10.1039/C7SC03267F
[67] "Non-Covalent Interactions in Molecular Crystals." G. Beran, Y. Heit, and J. Hartman. In Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications. Edited by A. Otero de la Roza and G. DiLabio. Elsevier. ISBN: 978-0-12-809835-6 DOI: 10.1016/B978-0-12-809835-6.00012-8
[66] "Measuring and modeling highly accurate 15-N chemical shift tensors in a peptide." S Soss, P. Flynn, R. Iuliucci, R. Young, L. Mueller, J. Hartman, G. Beran, and J. Harper. ChemPhysChem. 18, 2225-2232 (2017). DOI: 10.1002/cphc.201700357
[65] "Porous Materials: Designed and Then Realized." G. Beran. Nature Mat. 16, 602-604 (2017). DOI: 10.1038/nmat4913. Click here for free access.
[64] "Leveraging Electron Transfer Dissociation for Site Selective Radical Generation: Applications for Peptide Epimer Analysis." Y. Lyon, G. Beran, and R. Julian. J. Am. Soc. Mass Spectrom. 28, 1365-1373 (2017). DOI: 10.1007/s13361-017-1627-x
[63] "Fast Divide-and-Conquer Scheme for Evaluating Polarization in Classical Force Fields." D. Nocito and G. Beran. J. Chem. Phys. 146, 114103 (2017). DOI: 10.1063/1.4977981
[62] "Averaged Condensed Phase Model for Simulating Molecules in Complex Environments." D. Nocito and G. Beran. J. Chem. Theory Comput. 13, 1117-1129 (2017). DOI: 10.1021/acs.jctc.6b00890
2016
[61] "Predicting molecular crystal properties from first principles: Finite-temperature thermochemistry to NMR crystallography." G. Beran, J. Hartman, and Y. Heit. Acc. Chem. Res. 49, 2501-2508 (2016). DOI: 10.1021/acs.accounts.6b00404 Open Access.
[60] "Enhanced NMR discrimination of polymorphic pharmaceutically relevant crystals through ab initio fragment-based chemical shift predictions." J. Hartman, G. Day, and G. Beran. Cryst. Growth Des. 16, 6479-6493 (2016). DOI: 10.1021/acs.cgd.6b01157 Open Access.
[59] "Benchmark fragment-based 1-H, 13-C, 15-N and 17-O chemical shift predictions in molecular crystals." J. Hartman, R. Kudla, G. Day, L. Mueller, and G. Beran. Phys. Chem. Chem. Phys. 18, 21686-21709 (2016). DOI: 10.1039/C6CP01831A Open Access.
[58] "Crystal structure of the meta-stable intermediate in the photomechanical, crystal-to-crystal reaction of 9-tertbutyl anthracene ester." C. Yang, L. Zhu, R. Kudla, J. Hartman, R. Al-Kaysi, S. Monaco, B. Schatschneider, G. Beran, C. Bardeen, and L. Mueller. CrystEngComm 18, 7319-7329 (2016). DOI: 10.1039/C6CE00742B
[57] "How important is thermal expansion for predicting molecular crystal structures and thermochemistry at finite temperatures?" Y. Heit and G. Beran. Acta Cryst. B 72, 514-529 (2016). DOI: 10.1107/S2052520616005382. Part of a special issue on crystal structure prediction.
[56] "Structural switching in self-assembled metal-ligand helicate complexes via ligand-centered reactions." L. Holloway, H. McGarraugh, M. Young, W. Sontising, G. Beran, and R. Hooley. Chem. Sci. 7, 4423-4427 (2016). DOI: 10.1039/C6SC01038E Open Access.
[55] "Modeling polymorphic molecular crystals with electronic structure theory." G. Beran. Chem. Rev. 116, 5567-5613 (2016). DOI: 10.1021/acs.chemrev.5b00648 Open Access.
[54] "Predicting finite-temperature properties of crystalline carbon dioxide from first principles with quantitative accuracy." Y. Heit, K. Nanda, and G. Beran. Chem. Sci. 7, 246-255 (2016). DOI: 10.1039/C5SC03014E Open Access.
2015
[53] "Building blocks for bioinspired electrets: molecular-level approach to materials for energy and electronics." J. Larsen, E. Espinoza, J. Hartman, C.-K. Lin, M. Wurch, P. Maheshwari, R. Kaushal, M. Marsella, G. Beran, and V. Vullev. Pure Appl. Chem. 87, 779-792 (2015). DOI: 10.1515/pac-2015-0109
[52] "Reliable prediction of three-body intermolecular interactions using dispersion-corrected second-order Møller-Plesset perturbation theory." Y. Huang and G. Beran. J. Chem. Phys. 143, 044113 (2015). DOI: 10.1063/1.4927304
[51] "Fragment-based 13-C nuclear magnetic resonance chemical shift predictions in molecular crystals: An alternative to planewave methods." J. Hartman, S. Monaco, B. Schatschneider, and G. Beran. J. Chem. Phys. 143, 102809 (2015). DOI: 10.1063/1.4922649
[50] "Benchmark calculations of three-body intermolecular interactions and the performance of low-cost electronic structure methods." J. Rezac, Y. Huang, P. Hobza, and G. Beran. J. Chem. Theory Comput. 11, 3065-3079 (2015). DOI: 10.1021/acs.jctc.5b00281
[49] "Converging nuclear magnetic shielding calculations with respect to basis and system size in protein systems." J. Hartman, T. Neubauer, B. Caulkins, L. Mueller, and G. Beran. J. Biomol. NMR 62, 327-340 (2015). DOI: 10.1007/s10858-015-9947-2
[48] "High fidelity sorting of remarkably similar components via metal-mediated assembly." L. Holloway, M. Young, G. Beran, and R. Hooley. Chem. Sci. 6, 4801-4806 (2015). DOI: 10.1039/C5SC01689D
[47] First Reaction: "Compressive sensing in quantum chemistry: A little computation goes a long way." G. Beran. ACS Cent. Sci. 1, 14-15 (2015). DOI: 10.1021/acscentsci.5b00062 Invited First Reaction article discussing compressive sensing in quantum chemistry.
[46] Highlight: "A new era for ab initio molecular crystal lattice energy prediction." G. Beran. Angew. Chem. Int. Ed. 54, 396-398 (2015). DOI: 10.1002/anie.201409823. Invited Highlight article on the sub-kJ/mol accuracy obtained for the benzene crystal lattice energy.
[45] "Advances in molecular quantum chemistry contained in the Q-Chem 4 program package." Yihan Shao et al. Mol. Phys. 113, 184-215 (2015). DOI: 10.1080/00268976.2014.952696
2014
[44] "Fragment-based electronic structure approach for computing nuclear magnetic resonance chemical shifts in molecular crystals." J. Hartman and G. Beran. J. Chem. Theory Comput. 10, 4862-4872 (2014). DOI: 10.1021/ct500749h
[43] Editorial: "Calculation of Complex Bio- and Organic Systems: From Ground-State Reactivity and Spectroscopy to Excited-State Dynamics." A. Dreuw, G. Beran, and J. Neugebauer. ChemPhysChem 15, 3139-3140 (2014). DOI: 10.1002/cphc.201402644. Editorial for a special themed issue organized by the authors.
[42] "Exploiting space-group symmetry in fragment-based molecular crystal calculations." Y. Heit and G. Beran. J. Comput. Chem. 35, 2205-2214 (2014). DOI: 10.1002/jcc.23737
[41] "Dipole-mediated rectification of intramolecular photoinduced charge separation and charge recombination." D. Bao, S. Upadhyayula, J. Larsen, B. Xia, B. Georgieva, V. Nunez, E. Espinoza, J. Hartman, M. Wurch, A. Chang, C.-K. Lin, J. Larkin, K. Vasquez, G. Beran, and V. Vullev. J. Am. Chem. Soc. 136, 12966-12973 (2014). DOI: 10.1021/ja505618n
[40] "Achieving high-accuracy intermolecular interactions by combining Coulomb-attenuated second-order Møller-Plesset perturbation theory with coupled Kohn-Sham dispersion." Yuanhang Huang, Matthew Goldey, Martin Head-Gordon, and Gregory Beran. J. Chem. Theory Comput. 10, 2054-2063 (2014). DOI: 10.1021/ct5002329
[39] "Accurate molecular crystal modeling with fragment-based electronic structure methods." Gregory Beran, Shuhao Wen, Kaushik Nanda, Yuanhang Huang, and Yonaton Heit. in Prediction and Calculation of Crystal Structures: Methods and Applications edited by A. Aspuru-Guzik and S. Atahan-Evrenk, Springer, 59-93 (2014). DOI: 10.1007/128_2013_502
2013
[38] "What Governs the Proton-Ordering in Ice XV?" Kaushik Nanda and Gregory Beran. J. Phys. Chem. Lett. 4, 3165-3169 (2013). DOI: 10.1021/jz401625w
[37] "Communication: Constructing an implicit quantum mechanical/molecular mechanics solvent model by coarse-graining explicit solvent." Kelly Theel, Shuhao Wen, and Gregory Beran. J. Chem. Phys. 139, 081103 (2013). DOI: 10.1063/1.4819774
[36] "Accelerating MP2C dispersion corrections for dimers and molecular crystals." Yuanhang Huang, Yihan Shao, and Gregory Beran. J. Chem. Phys. 138, 224112 (2013). DOI: 10.1063/1.4809981
[35] "Boron carbides as efficient, metal-free visible-light-responsive photocatalysts." Jikai Liu, Shuhao Wen, Yang Hou, Fan Zuo, Gregory Beran, and Pingyun Feng. Angew. Chemie. Int. Ed. 52 3241-3245 (2013). DOI: 10.1002/anie.201209363. Selected as a "Hot Paper" by the journal editors.
[34] "Visible-light-responsive copper (II) borate photocatalysts with intrinsic midgap states for water splitting." Jikai Liu, Shuhao Wen, Xiaoxin Zou, Fan Zou, Gregory Beran, and Pingyun Feng. J. Mater. Chem. A. 1, 1553-1556 (2013). DOI: 10.1039/c2ta00522k
2012
 [33] "Prediction of organic molecular crystal geometries from
MP2-level fragment quantum mechanical/molecular mechanical
calculations." Kaushik Nanda and Gregory
Beran.
J. Chem. Phys. 137, 174106 (2012). DOI: 10.1063/1.4764063
[33] "Prediction of organic molecular crystal geometries from
MP2-level fragment quantum mechanical/molecular mechanical
calculations." Kaushik Nanda and Gregory
Beran.
J. Chem. Phys. 137, 174106 (2012). DOI: 10.1063/1.4764063
[32] "Crystal polymorphism in oxalyl dihydrazide: Is empirical DFT-D accurate enough?" Shuhao Wen and Gregory Beran. J. Chem. Theory Comput. 8, 2698-2705 (2012). DOI: 10.1021/ct300484h
[31] Editorial: "Fragment and localized orbital methods in electronic structure theory." Gregory Beran and So Hirata. Phys. Chem. Chem. Phys. 14, 7559-7561 (2012). DOI: 10.1039/C2CP90072F. Editorial for a special themed issue focusing on the fragment methods and local correlation guest-edited by Beran and Hirata.
 [30] "Accidental degeneracy in crystalline aspirin: New
insights from high-level ab initio calculations." Shuhao Wen and
Gregory Beran.
Cryst. Growth. Des. 12, 2169-2172 (2012). DOI: 10.1021/cg300358n
[30] "Accidental degeneracy in crystalline aspirin: New
insights from high-level ab initio calculations." Shuhao Wen and
Gregory Beran.
Cryst. Growth. Des. 12, 2169-2172 (2012). DOI: 10.1021/cg300358n
[29] "Practical quantum mechanics-based fragment methods for predicting molecular crystal properties." Shuhao Wen, Kaushik Nanda, Yuanhang Huang, and Gregory Beran. Phys. Chem. Chem. Phys. 14, 7578-7590 (2012). DOI: 10.1039/C2CP23949C
[28] "Structures and energetics of electrosprayed uracil_n Ca^2+ clusters (n=14-4) in the gas phase." Elizabeth Gillis, Maria Demireva, Kaushik Nanda, Gregory Beran, Evan Williams, and Travis Fridgen. Phys. Chem. Chem. Phys. 14, 3304-3315 (2012). DOI: 10.1039/C1CP22984B2011
[27] "Accurate molecular crystal lattice energies from a fragment QM/MM approach with on-the-fly ab initio force-field parameterization." Shuhao Wen and Gregory Beran. J. Chem. Theory Comput. 7, 3733-3742 (2011). DOI: 10.1021/ct200541h
[26] "Vibrations of a chelated proton in a protonated tertiary diamine". Gregory Beran, Eric Chronister, Luke Daemen, Aaron Moehlig, Leonard Mueller, Jos Oomens, Andrew Rice, David Santiago-Dieppa, Fook Tham, Kelly Theel, Sepideh Yaghmaei, and Thomas Morton. Phys. Chem. Chem. Phys. 13, 20380-20392 (2011). DOI: 10.1039/C1CP22065A
[25] "Conductance switching in diarylethenes bridging carbon nanotubes." Khalid Ashraf, Nicholas Bruque, Jeremy Tan, Gregory Beran, and Roger Lake J. Chem. Phys. 134, 024524 (2011). DOI: 10.1063/1.3528118
2010
[24] "Predicting organic crystal lattice energies with chemical accuracy." Gregory Beran and Kaushik Nanda. J. Phys. Chem. Lett. 1, 3480-3487 (2010). DOI: 10.1021/jz101383z
[23] "Electrochemical reduction of quinones: Interfacing experiment and theory for defining effective radii of redox moieties." Duoduo Bao, Sangeetha Ramu, Antonio Contreras, Srigokul Upadhyayula, Jacob Vasques, Gregory Beran, and Valentine Vullev. J. Phys. Chem. B. 114, 14467-14479 (2010). DOI: 10.1021/jp101730e
[22] "Spatially homogeneous QM/MM for systems of interacting molecules with on-the-fly ab initio force field parameterization." Ali Sebetci and Gregory Beran. J. Chem Theory Comput. 6, 155-167 (2010). DOI: 10.1021/ct900545v
2009
[21] "Conductance of a conjugated molecule with carbon nanotube contacts." Nicolas Bruque, Khalid Ashraf, Gregory Beran, Thomas Helander, and Roger Lake. Phys. Rev. B 80, 155455 (2009). DOI: 10.1103/PhysRevB.80.155455
[20] "The structure of the protonated adenine dimer by infrared multiple photon dissociation spectroscopy and electronic structure calculations." Khadijeh Rajabi, Kelly Theel, Elizabeth Gillis, Gregory Beran and Travis D. Fridgen. J. Phys. Chem. A 113, 8099-8107 (2009). DOI: 10.1021/jp9033062
[19] "Approximating quantum many-body intermolecular interactions in molecular clusters using classical polarizable force fields." Gregory Beran. J. Chem. Phys. 130, 164115 (2009). DOI: 10.1063/1.3121323
[18] "Computational investigation of thermochemistry and kinetics of steam methane reforming on Ni(111) under realistic conditions." Wayne Blaylock, Teppei Ogura, William Green and Gregory Beran J. Phys. Chem. C 113, 4898--4908 (2009). DOI: 10.1021/jp806527q
2008
[17] "Symmetry-breaking in benzene and larger aromatic molecules within generalized valence bond coupled cluster methods." Keith Lawler, Gregory Beran, and Martin Head-Gordon. J. Chem. Phys. 128, 024107 (2008). DOI: 10.1063/1.2817600
2007 and earlier:
[16] "Toward a comprehensive model of the synthesis of TiO2 particles from TiCl4." R.H. West, M.S. Celnik, O.R. Inderwildi, M. Kraft, G. Beran, and W.H. Green. Ind. Eng. Chem. Res. 46, 6147-6156 (2007).
[15] "First-Principles Thermochemistry for the Production of TiO2 from TiCl4." Richard West, Gregory Beran, William Green, and Markus Kraft. J. Phys. Chem. A, 111, 3560-3565 (2007).
[14] "On the nature of unrestricted orbitals in variational active space wave functions." Gregory Beran and Martin Head-Gordon. J. Phys. Chem. A, 110, 9915-9920 (2006).
[13] "Advances in methods and algorithms in a modern quantum chemistry program package." Yihan Shao, Laszlo Fusti Molnar, Yousung Jung, Joerg Kussmann, Christian Ochsenfeld, Shawn T. Brown, Andrew T.B. Gilbert, Lyudmila V. Slipchenko, Sergey V. Levchenko, Darragh P. ONeill, Robert A. DiStasio Jr, Rohini C. Lochan, Tao Wang, Gregory Beran, Nicholas A. Besley, John M. Herbert, Ching Yeh Lin, Troy Van Voorhis, Siu Hung Chien, Alex Sodt, Ryan P. Steele, Vitaly A. Rassolov, Paul E. Maslen, Prakashan P. Korambath, Ross D. Adamson, Brian Austin, Jon Baker, Edward F. C. Byrd, Holger Dachsel, Robert J. Doerksen, Andreas Dreuw, Barry D. Dunietz, Anthony D. Dutoi, Thomas R. Furlani, Steven R. Gwaltney, Andreas Heyden, So Hirata, Chao-Ping Hsu, Gary Kedziora, Rustam Z. Khalliulin, Phil Klunzinger, Aaron M. Lee, Michael S. Lee, WanZhen Liang, Itay Lotan, Nikhil Nair, Baron Peters, Emil I. Proynov, Piotr A. Pieniazek, Young Min Rhee, Jim Ritchie, Edina Rosta, C. David Sherrill, Andrew C. Simmonett, Joseph E. Subotnik, H. Lee Woodcock III, Weimin Zhang, Alexis T. Bell, Arup K. Chakraborty, Daniel M. Chipman, Frerich J. Keil, Arieh Warshel, Warren J. Hehre, Henry F. Schaefer III, Jing Kong, Anna I. Krylov, Peter M. W. Gill and Martin Head-Gordon. Phys. Chem. Chem. Phys., 8, 3172-3191 (2006).
[12] "The localizability of valence space electron-electron correlations in pair-based coupled cluster models." Gregory Beran and Martin Head-Gordon. Mol. Phys. 104, 1191-1206 (2006).
[11] "Second order correction to perfect pairing: An inexpensive electronic structure method for the treatment of strong electron-electron correlations." Gregory Beran, Steven R. Gwaltney and Martin Head-Gordon. J. Chem. Phys. 124, 114107 (2006).
[10] "A fast implementation of perfect pairing and imperfect pairing using the resolution of the identity approximation." Alex Sodt, Gregory Beran, Yousung Jung, Brian Austin, and Martin Head-Gordon. < J. Chem. Theory Comput. 2, 300-305 (2006).
[9] "Unrestricted perfect-pairing: The simplest wave-function-based model chemistry beyond mean field." Gregory Beran, Brian Austin, Alex Sodt, and Martin Head-Gordon. J. Phys. Chem. A 109, 9183-9192 (2005).
[8] "Fast electronic structure methods for strongly-correlated molecular systems." Martin Head-Gordon, Gregory Beran, Alex Sodt, and Yousung Jung. Journal of Physics: Conference Series--SciDAC 2005, 16, 233-242 (2005).
[7] "Search for stratospheric bromine reservoir species: Theoretical study of the photostability of mono-, tri-, and pentacoordinated bromine compounds." Timothy J. Lee, Cesar N. Mejia, Gregory Beran, and Martin Head-Gordon. J. Phys. Chem. A 109, 8133-8139 (2005).
[6] "Nitrogen activation via three-coordinate molybdenum complexes: Comparison of density functional theory performance with wave function based methods." David C. Graham, Gregory Beran, Martin Head-Gordon, Gemma Christian, Robert Stranger, and Brian F. Yates. J. Phys. Chem. A 109, 6762-6772 (2005).
[5] "Extracting dominant pair correlations from many-body wave functions." Gregory Beran and Martin Head-Gordon. J. Chem. Phys. 121, 78-88 (2004).
[4] "Local Correlation Methods." Martin Head-Gordon, Troy Van Voorhis, Gregory Beran and Barry Dunietz. Computational Science--ICCS 2003, Pt IV, Proceedings Lecture Notes in Computer Science 2660: 96-102, Springer-Verlag (2003) .
[3] "Approaching closed-shell accuracy for radicals using coupled cluster theory with perturbative triple substitutions." Gregory Beran, Steven R. Gwaltney, and Martin Head-Gordon. Phys. Chem. Chem. Phys. 5, 2488-2493 (2003).
[2] "Partitioning Techniques in Coupled Cluster Theory." Steven R. Gwaltney, Gregory Beran, and Martin Head-Gordon, in Fundamental World of Quantum Chemistry, Vol I, edited by E. Kryachko and E. Brandas, 433-457, Kluwer (2003).
[1] "Can coupled cluster singles and doubles be approximated by a valence active space model?" Gregory Beran, Steven R. Gwaltney, and Martin Head-Gordon. J. Chem. Phys. 117, 3040-3048 (2002).
Selected Review Articles, Editorials, and Press
Press: "Crystal structure prediction reaches new heights with axitinib." Highlight of our axitinib crystal structure prediction study. Chemistry World, Dec 9, 2025.
"Improved Description of Intra- and Intermolecular Interactions Through Dispersion-Corrected Second-Order Møller-Plesset Perturbation Theory" G. Beran, C. Greenwell, C. Cook, and J. Rezac. Acc. Chem. Res. 56, 3525-3534 (2023).
"Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials." G. Beran. Chem. Sci. 14, 13290-13312 (2023). Open Access.
Press: "Computational study predicts new high-pressure polymorph of ROY"" Highlight of our ROY crystal structure prediction study. Chemistry World, Jan 28, 2022.
"Calculating Nuclear Magnetic Resonance Chemical Shifts from Density Functional Theory: A Primer." G. Beran eMagRes. 8, 215-226 (2019).
Press: "Methanol polymorphs predicted with unprecedented accuracy." Highlight of our methanol phase diagram prediction article. Chemistry World, May 10, 2018.
Press: "What do Lego® bricks and crystal structures have in common?" Our article predicting the methanol phase diagram was the ChemSci Pick of the Week for May 2, 2018.
"Porous Materials: Designed and Then Realized." G. Beran. Nature Mat. 16, 602-604 (2017). Click here for free access.
"Modeling Polymorphic Molecular Crystals with Electronic Structure Theory." G. Beran Chem. Rev. 116, 5567-5613 (2016). Open Access.
"Predicting molecular crystal properties from first principles: Finite-temperature thermochemistry to NMR crystallography." G. Beran, J. Hartman, and Y. Heit. Acc. Chem. Res. 49, 2501-2508 (2016). Open Access.
First Reaction: "Compressive sensing in quantum chemistry: A little computation goes a long way." G. Beran. ACS Cent. Sci. 1, 14-15 (2015). Invited First Reaction article discussing compressive sensing in quantum chemistry.
Highlight: "A new era for ab initio molecular crystal lattice energy prediction." G. Beran. Angew. Chem. Int. Ed. 54, 396-398 (2015). Invited Highlight article on the sub-kJ/mol accuracy obtained for the benzene crystal lattice energy.
"Accurate molecular crystal modeling with fragment-based electronic structure methods." Gregory Beran, Shuhao Wen, Kaushik Nanda, Yuanhang Huang, and Yonaton Heit. in Prediction and Calculation of Crystal Structures: Methods and Applications edited by A. Aspuru-Guzik and S. Atahan-Evrenk, Springer, 59-93 (2014).
"Practical quantum mechanics-based fragment methods for predicting molecular crystal properties." Shuhao Wen, Kaushik Nanda, Yuanhang Huang, and Gregory Beran. Phys. Chem. Chem. Phys. 14, 7578-7590 (2012).