Overview
The Beran group uses computational quantum chemistry methods to understand and predict chemistry in complex systems. We are particularly interested in how organic molecules pack in the solid state and molecular crystal polymorphism, and how theoretical predictions can be coupled with experiment to solve challenging crystal structures. In the course of these studies, we often need to develop and implement new theoretical models and computational algorithms in order to make the studies computationally feasible.
Click on the tabs above to learn more about our research group, or take a look at some of our review articles:
- "Frontiers of molecular crystal structure
prediction for pharmaceuticals and functional organic materials."
G. Beran.
Chem. Sci. 14, 13290-13312 (2023). DOI: 10.1039/D3SC03903J
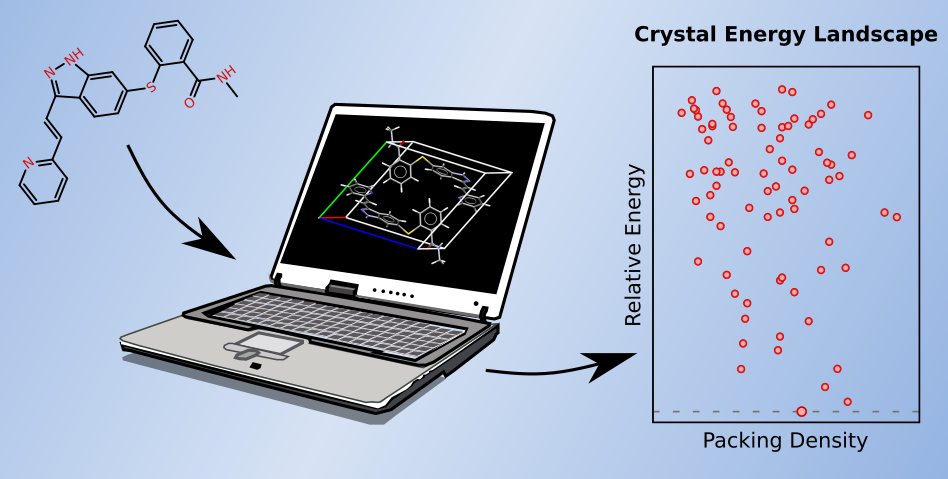
- "Improved Description of Intra- and Intermolecular
Interactions Through Dispersion-Corrected Second-Order Møller-Plesset
Perturbation Theory" G. Beran, C. Greenwell, C. Cook, and
J. Rezac.
Acc. Chem. Res. 56, 3525-3534 (2023).

- "Predicting molecular crystal properties from first principles:
Finite-temperature thermochemistry to NMR crystallography."
G. Beran, J. Hartman, and Y. Heit.
Acc. Chem. Res., 49, 2501-2508 (2016).
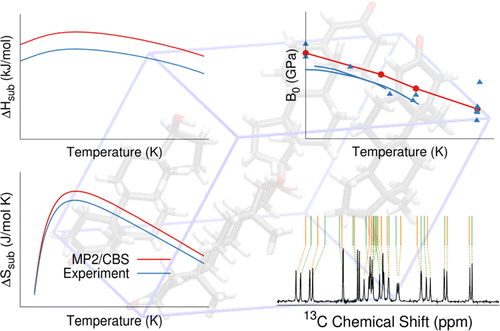
- "Modeling polymorphic molecular crystals with electronic structure theory." G. Beran. Chem. Rev. 116, 5567-5613 (2016).
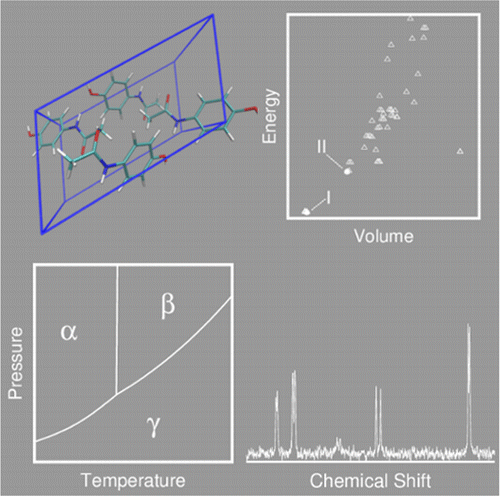
- "Modeling polymorphic molecular crystals with electronic structure theory." G. Beran. Chem. Rev. 116, 5567-5613 (2016).
People interested in joining the group should email Prof. Beran.
